| Coma and other disorders of consciousness |
| Jouvet M. Handbook of Clinical Neurology Vol.3. P. J. Vinken and G. W. Bruyn , eds. North-Holland Publishing Company. Amsterdam,(1969) |
Introduction
Coma is classically defined as loss of motility, sensation and consciousness with preservation of autonomic functions. Though not inaccurate, this definition is no longer considered as adequate. If loss of consciousness seems to be a necessary and sutficient symptom to talk of coma (it being understood that we are dealing with a lethargic state very different from sleep, in as much as the subject cannot be roused by strong external stimulation), it follows that loss of voluntary motility and sensation are natural consequences of the loss of consciousness and add no useful precision to the definition. On the other hand, autonomic disturbances almost invariably accompany a comatose state; this at least was the case prior to the introduction of modern resus citation methods. For these reasons, the definition of coma rests on the definition of consciousness. But as Jackson himself has stressed, consciousness is very difficult to define. We refer the reader to the following recent papers concerning the problem of definition of consciousness in neurophysiological terms (Fessard 1954; Alajouanine 1957; Walshe 1957; Eccles 1966; Adrian 1966; MacKay 1966). However, modern methods of investigation and in particular neurophysiological clinical methods applied to the study of prolonged comas (the symptomatology of which is less variable than that of acute comas) and the importance attached to the dynamic and dialectic concept of perceptivity * and of reactivity *, will lead to another definition borrowed from psychophysiology. According to this definition, consciousness is that central nervous process which gives significance to a stimulus from the external environment. We can thus understand by consciousness that function of the nervous system which is concerned with the perceptual experience of information from the environment and from our own body (Alajouanine 1957). The multiple disintegration of consciousness observed in coma can therefore be defined as the absence, in the patient, of objective clinical (or paraclinical) signs of appreciation of his environment. It is this behavioural deficiency that the clinician explores by simple means: failure to rouse a comatose patient by calling his name or applying a painful stimulus.
* The terms 'perceptivity' (perceptivité) and 'reactivity' (réactivité) are adjectives applied to central mechanisms which are necessary for perception and reaction.
Physiological Basis of Coma (introductory remarks)
The physiopathology of coma poses three main problems :
(1) Which structures are required for the conscious appreciation of a stimulus? In terms of neuro physiology, we may say that conscious appreciation depends on the integrity of cortical structures, provided that the waking system of the brainstem is itself functionally intact.
(2) Conscious appreciation is not a stable and permanent phenomenon. It is subject to a physiological dissolution by reason of the presence of sleep "systems" which periodically counteract the waking system. Now, do the sleep systems play a part in the production of a comatose state ? The answer would appear to be "no" as organic lesions of the sleep systems in animals cause no apparent disturbance of consciousness.
(3) The last problem is histopathological: what are the exact mechanisms by which neurological lesions lead to a comatose state ?
Reactivity and perceptivity : it is important to make a distinction between the two basic concepts lead of reactivity and perceptivity (Alajouanine 1957) as we shall later rely on these notions to assess the depth of coma in man and animals.
Reactivity brings into play mechanisms which are present since birth, the seat of which is subcortical; these are activated via the telereceptors (the eyes and ears) or via the nociceptive stimuli.The arousal reaction, the orienting response (rotation of the head towards the source of an auditory stimulus) and the facio-vocal reactions to pain are all part of the reactivity complex.
Perceptivity on the other hand implies the response of nervous mechanisms acquired by learning. It is a response to stimuli of a more complex nature (words, gestures, writing) or to simpler ones (elementary conditioning in the case of blinking response to a threat). We now know that these responses, which depend upon recent and long-term memory, require a certain degree of cortical integration . The integrity of the cerebral cortex, however, is a necessary but not a sufficient condition to ensure normal perceptivity. We must go further, therefore, in our neurophysiological analysis of the nervous mechanisms which are essential for the awareness of an external event.
Nervous structures necessary for consciousness
A priori, failure to "perceive" a signal may result theoretically from the following lesions (Fig. 1):
- a lesion of the ascending pathways specifically responsible for the conduction of stimuli from the external environment to the "integrating central structures";
- a lesion of the efferent pathways responsible for providing external evidence that the signal has been integrated (i.e. via muscular or vegetative effectors);
- finally, a lesion affecting the integrating central structures.
Neurophysiology has shown that the first two hypotheses do not apply in the great majority of cases. Indeed, lesions simultaneously affecting the afferent pathways on both sides are rare; besides, in animals, experimental de-afferentation of the various receptors, or even transection of the specific lateral afferent pathways in the brainstem (without damaging the central paft of the tegmentum) do not lead to a comatose state. The animal is still capable of reacting correctly to a number of conditioned reflexes (Sprague et al. 1963). Also, if a general de-efferentation may sometimes result in a clinical picture of coma, a closer examination would usually show that normal perception is preserved. This is the case in patients suffering from advanced tetanus, and are kept under artificial respiration after intense and prolonged curarization. These patients may at first sight appear comatose as they are unable to show objectively by means of movements their perception of external stimuli, and yet they are fully aware of their surroundings. We tested this by showing our curarized patients various cards (ace of hearts, king of spades) and asking them to identify the cards later in a shuffled pack of cards. When they recovered and curarization was stopped, they were perfectly capable of recognizing the cards which had previously been shown to them. These cases are exceptional as there is no clinical test that enables us to assess the state of consciousness of a curarized patient. It is possible that an electroencephalogram by showing an arousal reaction, might provide important, if not decisive information.
Even though it may appear to be of purely theoretical interest, this question is of prime importance. Some lesions of the brainstem, in particular lesions of the ventral part of the mesencephalic or pontine tegmentum, can in fact involve the extrapyramidal system, and prevent certain voluntary movements, as well as cause a motor de-efferentation. At this point, we would like to mention an observation by Lhermitte et al. (1963). A patient with a lesion of the mesencephalic reticular formation presented the clinical picture of deep coma. She had bilateral ptosis, which gave the impression that she was in a state of permanent sleep, and failed to respond even to strong stimuli. It was noticed, however, that she was still able to move her wrists slightly. Due to these movements of the wrist, it was possible to establish a code by means of which she was able to answer complicated questions in a simple manner. This fact leads to two practical points worth remembering when one is faced with patients presenting a state of "akinetic mutism". First, one should always have recourse to all possible paraclinical examinations (including polygraphy) before deciding that a patient has lost all perception. Secondly, one should always remember that a patient who is akinetic and speechless, may still understand all that is being said around him.
It would appear therefore that neither a subtotal nor a total de-efferentation can (alone) result in complete loss of perception. This is why disturbances of consciousness seem to be connected with an organic or functional lesion of the integrating central structures. These structures belong to two main groups. The first group includes the ascend ing activating reticular system, the normal functioning of which is essential for waking, but is not sufficient to insure normal perceptivity. The second group includes the thalamocortical integration system, the integrity of which is necessary for normal perceptivity.
The ascending reticular activating system (ARAS) and the arousal reaction.
The brainstem reticular formation includes the areas of grey matter in the bulbar tegmentum, the pons and the mesencephalon, but excluding the cranial nerve nuclei, and the relay centres of the cerebellar system. It is continuous at the bulbospinal junction with the spinal reticular formation. Anteriorly, its limits are not clearly defined, and in the sub thalamic area it is continuous with the diffuse thalamic system. Thus the reticular formation appears as a complex collection of neurones which serves as a converging point for signals from the external and the internal environments, and is capable of exercising a dynamogenic effect on the electrical activity of the cortex, and on the motor and vegetative efferent system.
Since 1949, thanks largely to the work of Moruzzi, Magoun and their school (Lindsley et al . 1950; French and Magoun 1952) it has been shown that after destruction of the mesencephalic reticular formation, an animal remains immobile and "comatose". Sensory stimuli, auditory or painful, fail to clinically elicit the arousal reaction. The electrical activity recorded from the cortex is similar to a sleep-recording, and with the exception of olfactory stimuli, all forms of sensory stimulation fail to elicit a reaction. Excitation of the reticular formation in sleeping animals confirms its activating influence: when high frequency excitation is applied, the animal wakes up, opens its eyes, and there is mydriasis and increase in muscle tone, etc.; in some cases, the animal will give an impression of attention. At the same time, there is a rapid, low voltage EEG cortical activity which is characteristic of the "arousal reaction".
This work led to the discovery in the brainstem of a system of neurones which is now regarded as the basis of the waking mechanism. Further work has shown that the structures responsible for the maintenance of the waking state are situated in the posterior diencephalon and at the mesodiencephalic junction, while the cell groups responsible for cortical activation are localized in the posterior mesencephalon and the anterior part of the nucleus reticularis pontis oralis (Rossi and Zanchetti 1956).
Cortex, perceptivity and learning.
It is now established that the cortical centres are responsible for the nerve connections acquired during the history of the organism. Thus, after decortication, either widespread or limited to a specific area, all the possibilities of conditioning, achieved in animals by means of complex stimuli, are lost. This state is comparable in man to visual, auditory, somesthetic agnosia resulting from lesions either of a specific cortical area or of the nonspecific adjacent areas of integration (Ajuriaguerra and Hécaen 1960).
It is necessary at this point to sum up briefly the present data concerning the structures responsible for the facio-vocal reactions to pain. Indeed, investigation of reactions to painful stimuli should be an integral part of the examination of a comatose patient, as it serves to test some of the essential mechanisms of reactivity.
Structures responsible for responses to painful stimuli.
We know that a painful stimulus activates a complex afferent system, the organisation and integration centres of which are only now being partly elucidated. We can accept the view of Bard and Mountcastle (1948) according to which the neocortex, the cingulate cortex, the amygdaloid nucleus and the pyriform lobe correspond to zones of the inhibition of pain and anger reactions. Their influence would be transmitted as far down as the brainstem by way of a circuit similar to the amygdaloid pathway. They suggest the presence, in addition, of a direct extra-amygdaloid pathway via which the neocortex might exert a facilitatory influence on the mesencephalic centres.
The mesencephalic structures where the facial and vocal components of the pain reaction are integrated are situated in the central part of the brainstem, within the peri-aqueductal grey matter. Destruction of this area in animals results in complete loss of vocal or expressive reactions following a painful stimulus (Adametz and O'Leary 1959; Kelly et al. 1946), while excitation of this same area provokes intense vocal and facial reactions. The vegetative responses to pain (pupillary, respiratory, cardiac vasomotor) are elaborated in the inferior part of the brain stem; they result from mechanisms originating in the mesencephalic, rhombencephalic and bulbar areas. The spinal component may suffice when a limb is withdrawn from a painful stimulus applied to the skin; this is the classical flexor reflex.
And finally, although no absolute correlation exists between the disturbances of perceptivity and reactivity on the one hand, and of muscle tone on the other, in most cases a clinical correlation is found between lesions of the integrative structures and those affecting the centres controlling muscle tone.
Neurophysiological basis of abnormalities of posture.
In man, disturbances of posture, which are char acterised by the rigidity
of decortication and de cerebration, depend largely on the multisynaptic
systems of the brainstem. And, as we know, the systems responsible for
the waking reaction are closely associated with the supraspinal centres
controlling muscle tone. For that reason, we would like to summarize some
of the neurophysiological experimental data concerning the regulation
of muscle tone (Granit 1957; Jung and Hassler 1960; Rushworth 1960). Muscle
tone depends on the simultaneous control of the facilitatory and inhibitory
supraspinal systems, which act on the stretch reflex.
The inhibitory influences, independent of the pyramidal system, follow
a corticobulboreticular system which originates in the suppressive areas
of the cortex, or a multi-relay system at the level of the striate nuclei,
or again they may depend on the anterior lobe of the cerebellum. Of the
supra spinal control systems, the bulbar inhibitory reticular system is
one of the most dominant. Its influence reaches down to the spinal motoneurones
by way of the reticulospinal tract.
The facilitatory influences, which act on the spinal level, come from the vestibulospinal tracts (probably unimportant in man), the middle lobe of the cerebellum, the pyramidal tract, and especially from the facilitatory reticular formation which is situated in the mesencephalic tegmentum, and lies very close to the inhibitory reticular formation at tne pons.
Spasticity appears whenever a lesion affects the inhibitory centres and leaves the facilitatory system to act unopposed on the spinal stretch reflexes. According to this classical conception, in the presence of a cortical lesion, the inhibitory reticular formation, though not organically damaged, undergoes a sort of "isolation dystrophy" which prevents its inhibitory action on muscle tone, and this results in spasticity. It has been shown in fact (Jouvet et al. 1961) that in the cat and in man, the reticular formation continues to have a periodic inhibitory influence in certain stages of sleep (paradoxical sleep). Thus rigidity resulting from suprapontine lesions may still be under a periodic inhibitory influence during sleep.
Thus, according to the various types of experimental lesions studied in animals, we may consider the following schematic pictures:
Total decortication in the cat (in direct opposition to what happens in man) is accompanied by minimal changes in muscle tone. On the other hand, perceptivity is completely absent: the "blinking" reflex is lost, although the light reflex persists, and the animal is unable to move to wards his food. One orientation reaction remains: startling, turning the ears and rotating the head towards a noise. Finally, the ability to learn is lost, and no conditioned reflex can be demon strated. Reactivity however remains normal: the arousal reaction is present, and the animal can be aroused from its sleep by an acoustic stimulus. Finally, the facio-vocal reactions to pain are exaggerated and result in an expression akin to that of "sham rage".
Destruction of the ascending reticular actitvating system. Here, even though the cortex is intact, perception is completely abolished, while reactivity is severely impaired. The animals also show signs of severe muscle tone disturbances (resulting in more typical cases, in a clinical picture of decerebrate rigidity in extension). The eyes are closed; the pupils are constricted; auditory and painful stimuli fail to provoke an arousal or an orientation reaction. Finally, cortical activity consists entirely of slow waves and remains unchanged by all sensory stimuli with the exception of olfactory stimuli.
In addition to these two generalized syndromes, there are those due to more localized lesions. One of these is experimental akinetic mutism resulting from lesions of the median structures situated in the peri-ependymal grey matter and the medial thalamic nuclei. The disturbances of perceptivity are more apparent than real, and appear to be due to damage to the motor effector mechanisms. The animal is still able to blink when threatened, but its immobility makes motor conditioning difficult to achieve. The waking reaction is normal, but facio-vocal reactions to pain are completely absent. There is also a generalized disturbance of muscle tone which in the more typical cases may result in catatonia.
Finally, a more generalized lesion (most com monly seen after prolonged anoxia) affecting the cortex, the mesencephalic reticular formation, the pontine reticular formation and other more caudal structures will lead to a state of brain death or even to the picture of "isolated heart-lung preparation", on condition that artificial respiration is applied. This clinical state is the ultimate stage of coma: it is characterized by complete absence of perceptivity and of reactivity, and by total cerebral electrical silence.
Periodic physiological dissolution of consciousness: sleep and coma
Etymologically, coma means sleep, and for a long time coma has been regarded as a very deep sleep. One is then entitled to wonder whether some forms of coma are not due to the exaggeration of the normal sleep mechanism. This seems unlikely for the following reasons which we shall discuss briefly:
(1) Lesions of the sleep systems in animals result in a state of prolonged insomnia, without disorders of perceptivity.
(2) Stimulation of the sleep systems will induce a reversible sleep, but never causes prolonged loss of consciousness.
Structures responsible for the states of sleep. Two forms of sleep are recognized: one which is characterized by slow, or synchronized, cortical activity, with maintenance of some muscle tone: "slow sleep"; the second is accompanied by a higher frequency cortical activity of low voltage (similar to that in the waking state), complete loss of muscle tone, and rapid eye movements: this is "paradoxical sleep" (Jouvet 1967). This second sleep occurs periodically during physiological sleep and constitutes about 20 % of normal sleeping time. It corresponds in man to oneiric activity.
It has been shown that these states of sleep are the result of an active process of inactivation of the waking system. The theory according to which sleep is due to passive inactivation of the activating reticular system has now been abandoned. It is in fact possible to induce prolonged insomnia by destroying the hypnogenic systems. These are situated in the lower part of the brainstem (pons and medulla) and there appear to be two distinct systems: one, situated at the level of the nuclei of Raphé, which is associated with slow sleep, and one situated in the laterodorsal part of the pontine tegmentum which is associated with paradoxical sleep.
Destruction of these systems causes elective suppression of the sleeping states. But this lesion, which causes considerable prolongation of the waking state, induces no disturbances of perceptivity or of reactivity in animals.
It is possible on the other hand, to bring on one or the other form of sleep by stimulating, under well defined conditions either the sleep centres or some of their afferent or efferent connections. The sleep induced in this way is always short-lasting and is always easily reversed by external stimu lation. Prolonged loss of consciousness has never been induced experimentally in animals by stimulation of the sleep systems.
Although a priori it may seem possible that certain inflammatory processes (Von Economo's epidemic encephalitis) might lead to chronic excitation of the hypnogenic structures, it seems unlikely that hypersomnia and encephalitic le thargy are due to lesions of these structures. In animals at least, reversible hypersomnia and irre versible coma have always been induced by lesions of the waking system.
This observation leads to the following corollaries which may be of some practical interest:
- (irreversible) coma must not be identified with sleep;
- some forms of coma (due to an organic or functional lesion of the waking system) can there fore be actively modulated by activation of the two hypnogenic systems which, under normal conditions, are responsible for slow and paradoxical sleep respectively. This modulation ex plains the various degrees of loss of perceptivity or reactivity observed in certain patients at different times, and which might be due to the fact that the few structures concerned with waking spared by the lesion are periodically submitted to an active process of inactivation from the sleep systems. Finally, total muscular atony such as is found in paradoxical sleep, may occur periodically in states of rigidity due to decortication or to decerebration. This explains the fluctuations in muscle tone encountered during continuous poly graphic recordings (Jouvet et al. 1961) (Fig.2).
From experimental to clinical neurophysiology
If the nervous structures responsible for waking and for the various types of sleep in animals (and especially the cat) are fairly well-known, anatomoclinical observations and neurophysiological clinical investigations seem to indicate that in man the equivalent structures may present a different organization .
In 1960, Jefferson drew a map of the areas of the brain which he believed, from his neurosurgical experience, to be necessary for the preservation of consciousness (critical point). The mapped region extends from the pons, through the mesencephalon and diencephalon, as far as the telencephalon. It appears however that the localization of structures responsible for maintaining the waking state must be based on the lowest lesion that will produce a prolonged loss of consciousness, for coma produced by a higher lesion can be due to the interruption of ascending neurones originating from the more caudal activating centres.
Rossi and colleagues (1964, 1965), after study ing their own personal observations, as well as those they found in the literature, have localized with more precision the regions of the brainstem which are vItal. in maintenance of a waking state: bulbar lesions and lesions of the caudal part of the pons cause no loss of consciousness (Fig.3) even if decerebrate rigidity is present (Halsey and Downie 1966), while lesions of the upper part of the pons and especially of the lower end of the mesencephalon are almost always associated with coma. In these cases, however, the EEG tracing may be normal even though no reactions appear on the tracing (dissociation between EEG and behaviour) (Chatrian et al. 1964; Loeb and Poggio 1953; Kaada et al. 1961).
Finally, lesions of the cephalic end of the brainstem and of the posterior part of the diencephalon always result in a picture of deep coma, both clinically and electroence phalographically (slow, non-reactive EEG). It appears therefore that the most caudal lesion of the brainstem capable of producing coma is one situated at the junction of the mesencephalon and diencephalon .
Alema et al. (1966) have also used a functional method to identify and localize the structures responsible for maintaining the waking state (and therefore normal consciousness). In man, injection of Amytal into the vertebral artery causes in activation of the lower brainstem and takes the form of bilateral palsy of the 12th, 7th, 6th, 5th, 4th and 3rd cranial nerves, as well as loss of autonomic functions of the 3rd (mydriasis and absence of light reflex); the sensory part of the 5th nerve is also affected. But in spite of this functional inactivation of the lower brainstem, the subjects show no loss of perception and continue to react normally to auditory and visual stimuli (Fig.3).
So, the combined results of anatomoclinical studies and of injection of barbiturates into the vertebral artery indicate that, in man,the neurones responsible for maintaining the waking state (and thus indirectly responsible for normal perceptivity) are situated at the junction of the mesencephalon and diencephalon.
Physiopathology of nervous lesions responsible for coma
Coma often results from diffuse cerebral lesions (e.g. of vascular, neoplastic or infectious origin) which are difficult to reproduce experimentally. By means of experimental head injuries, however, it has been possible to make a detailed study of the mechanisms by which primary or secondary lesions may lead to loss of consciousness (Denny Brown and Russel 1941; Holbourn 1943; Row botham 1949; Gurdjian and Webster 1958; Ward 1958; Foltz et al. 1953; De Morsier 1956).
Primary disorders
Mechanical displacement to the brain, such as occurs in a head injury,
results in functional and anatomical lesions proportional to the severity
of the shock.
Cerebral concussion is a transitory and reversible nervous reaction of sudden onset following physical trauma of brief duration and sufficient severity, and characterized by progressive recovery. It seems that the transitory traumatic paraIysis of the neurones might be due primarily to damage inflicted on the membranes (but with no detectable histological lesion). It is characterized by a brief loss of consciousness, with amnesia, and is never associated with prolonged coma.
When the head injury is severe enough, the disrupting forces may cause displacement of the whole cerebrum. The lesions will then depend on the degree of displacement, on which parts are displaced, and on the direction of the disrupting forces in relation to that of the nerve fibres. Such lesions are accompanied by histological changes ranging from loss of myelin to diffuse degeneration of white matter of the cerebrum (Fig.4) which are characteristic of traumatic encephalopathy as described by Strich (1956). In certain cases, the shock of the hemispheres hitting against the falx cerebri, or the lateral displacement of the hemispheres submit the corpus callosum to stretching forces which are responsible for contusions inside the corpus callosum at its points of insertion.
Foci of cerebral contusion are one of the anato mical characteristics of a brain after a head injury, and they have been described by a number of authors (Symonds 1949; Peters 1955; Gurdjian et al. 1955; Wertheimer and Descotes 1961). These histological changes, usually necrosis or haemorrhage, occur more frequently at certain points, suggesting that they are the result of mechanical forces, either from direct shock or by the classical contre-coup: the lesions are then found at the opposite pole from the point of impact, on the inferior aspect of the frontal lobe, at the temporal poles. In a few cases, the primary lesions may affect the brainstem directly (Jefferson 1952; French 1952; Mansuy et al. 1955).
Secondary lesions
There are two types of anoxic lesions: ischaemic anoxia in the case of prolonged shock with hypotension, and anoxic anoxia from lesions of the respiratory centres (apnoea, Cheyne-Stokes) or from upper respiratory tract involvement (the classical tracheobronchial congestion which tracheotomy has almost abolished). These lesions are now more clearly understood following the work of numerous investigators (Grenell 1946; Malamud and Haymaker 1947; Courville 1950, 1953; Scholtz 1953; Meyer and Denny-Brown 1955; Meyer 1956; Zeman and Youngue 1957; Mandel and Berry 1959).
It has been clearly established that the cerebellar cortex and cerebral cortex (more particularly the frontal cortex and hippocampus) are the most sensitive to anoxia, the next most sensitive being the pallidum, which is a common site for anoxic lesions (Scholtz 1953).
Finally, severe anoxia may lead to massive aseptic necrosis of the encephalon (Fig.5). The appearance is one of generalized necrosis primarily affecting the nerve cells and usually sparing the axons. The cerebral cortex, the cerebellum, the central grey nuclei and the brainstem are all affected. An important fact is that in the midst of this massive lesion of nervous tissue, there is no glial reaction (Bertrand et al. 1959; Mollaret et al. 1959; Trillet 1961). The picture of massive aseptic necrosis of the brain is found in the latest stages of coma, mainly in "brain death".
Cerebral oedema may also occur and cause the process known as necrosis of oedema since the works of Jakob (1913) and of Hallervorden (1939). It is characterized by foci of myelin disintegration affecting the subcortical white matter, and some times even by "slits of disintegration".
In certain cases, contusion, distortion, anoxia and oedema all combined may cause the rather rare pathological appearance of diffuse, traumatic degeneration of cerebral grey matter characterized by the disappearance of cells in the cortex, the thalamus, the putamen and the red nucleus (Denst et al. 1958).
The preceding data show the importance of anoxia (or hypoxia) in the development of certain secondary organic lesions leading to coma. By modern methods of investigation (by using, e.g. krypton-85) it has been shown that in certain comatose patients (in whom cerebral biopsy had revealed no abnormality) the blood supply and metabolism of the hemisphere could be reduced by up to 75% (Ingvar et al. 1964).
Aetiological classification of comas and of disturbances of conciousness of organic origin
The main causes of coma, which belong to numer ous fields of neurology, are summed up in Table 1.
TABLE 1 (After Silvermann 1963)
1 Cerebral vascular diseases
- (a) Vertebrobasilar or carotid thrombosis
- (b) Haemorrhage (cerebral or meningeal)
- (c) Embolism
- (d) Hypertensive encephalopathy
2 Head injuries
- (a) Concussion
- (b) Contusion, laceration, distortion
- (c) Haematomas (intracerebral, subdural, extradural)
3 Brain tumours
- (a) Gliomas
- (b) Meningiomas
- (c) Metastases
4 Infectious diseases
- (a) Encephalitis
- (b) Meningitis
- (c) Cerebral abscess
- (d) Diseases due to parasites
5 Degenerative diseases
- (a) Disseminated sclerosis
- (b) Schilder-Foix disease
- (c) Van Bogaert's sclerosing encephalitis
- (d) Senile and presenile dementias
- (e) Various others
6 Miscellaneous
- (a) Epilepsies of various origins and postcritical coma
Symptomatological classification of coma
Advances in rescuscitation methods (and especially the prevention of anoxia by tracheotomy) have made it possible for the majority of comatose patients to survive the acute stage during which autonomic disturbances often conceal some of the deficiency aspects of the loss of consciousness. For this reason, it is more interesting to study the chronic and prolonged cases of coma which are now more frequently encountered. Many attempts have been made at classifying the various "depths" of coma - namely, the classical one which distinguishes between obnubilation, torpor, light coma and coma carus. Most authors distinguish three or four stages or "depths" (Mansuy et al. 1955; Fischgold and Mathis 1959; Loeb 1958).
These classifications have the distinct advantage of simplicity, but they fail to distinguish between the signs of cortical damage and those of brain stem damage. Besides, the evolution of states of unconsciousness is often variable, and the fluctuations must be followed fairly closely as they are of great prognostic value and may influence the line of treatment. For this reason it is desirable to have a less rigid classification which would recognize a wider range of objective signs easy to elicit on simple physical examination. The following classification is based on the criteria of per ceptivity and of reactivity described earlier (Jouvet and Dechaume 1960).
The basis of this classification is the study of 40 unconscious patients (the duration of un consciousness ranging from 40 to 3700 days). The majority of these (17) were posttraumatic, the others resulted from encephalitis, vascular accident, and the outcome of neurosurgery.
In addition to these 40 cases of prolonged un consciousness, we have studied 14 cases of brain death, kept alive by artificial methods from 24 hours to 6 days (10 posttraumatic, 2 post operative, 2 vascular accident).
Perceptivity (P)
The following are the tests we selected :
In the first place, test the response to a written order: "close your eyes", "put out your tongue".
The second question is designed to test the pa tient's orientation in time and space: "do you know where you are ?", "do you know which day it is ? which month? which year?"
The third test consists of a simple mental calculation: "what is 8 x 7 -- 6 x 6 ?" Usually, the answer to 8 x 7 disappears long before that to 6 x 6. The fourth test consists in asking the patient to name ten flowers. Usually, during progressive "dissolution of consciousness", the patients can only name one flower, most often the rose.
The fifth test consists in studying the patient's ability to obey a spoken command: ask him to shut his eyes or to put his tongue out. (Asking him to open his eyes is pointless, as some patients will open their eyes in response to any auditory stimulus; this is the nonspecific reaction.)
In the sixth test, one watches for the presence of the blink reflex: this is carefully tested by suddenly moving a reflex hammer towards the root of the nose (the palm of the hand must not be used as air displacement may well trigger of the corneal reflex).
Thus (Table 2), depending on the responses to these different tests, it will be possible to place the patient in one of five categories, according to the disturbances of perception:
- P1 includes all the patients with no loss of con sciousness, patients who are "neurophysiologically" normal.
- P2 represents "obnubilation": these patients are disorientated in time and in space, and are unable to obey a written command. They can, however, pass all the other tests.
- P3 represents what was classically known as "torpor": they cannot give the name of flowers, and their understanding of the spoken word is poor. An order must be repeated many times before it is obeyed, and even then very slowly. But the blink reflex remains normal.
- P4 represents the last stage of cortical percept ion, where only the blinking response to a threat persists.
- P5 represents the stage where all perception is lost, indicating an organic or functional disturb ance of the cortical neurones.
Nonspecific reactions (R)
These are easily tested. It is essential to start by testing the audition to make certain that one is not dealing with a deaf patient: this is done by testing the cochleopalpebral reflex, where a loud noise causes blinking of the eyelid.
Orientation reaction:
if the patient has his eyes open, one stands by the side of the bed and makes a loud noise or calls his name: the orientation reaction, if present, will cause the patient to rotate first his eyes, and then his head towards the source of the noise.
Waking reaction:
if the patient keeps his eyes shut and appears to be "asleep", the same ma noeuvre is performed, and one watches for the opening of the eyes. If the eyes stay open, it is then possible to place the patient in one of three groups:
- R1 includes those patients who show a positive orientation reaction with their eyes open, and a positive waking reaction with their eyes shut.
- R2 includes those who have lost the orientation reaction with their eyes open, but can still open their eyes when challenged.
- R3 includes those where the waking functions of the brainstem have been lost.
Reaction to pain (D)
This is the next stage of the examination - it is judged according to three criteria (Fig. 6):
Facial expression must be observed carefully. It is usually a combination of upper facial move ment with wrinkling of the lids, sometimes with opening of the eyes, wrinkling of the forehead and especially a mimic of the lower part of the face with the grimace characteristic of pain: it is usually accompanied by a vocal reaction, a grunt, sometimes heard with difficulty in tracheotomized patients. It is also important to note if in certain patients a painful stimulus only elicits a waking reaction
Finally, the terminal stage of the reaction to pain is the withdrawal of the stimulated limb: pinching of a limb usually causes a reaction which may be obvious, or which is only perceptible in one muscle group.
On the basis of the reaction to pain, patients can again be divided into four groups
- D1 is the group where reaction is normal: there is the characteristic mimic, the cry, the waking reaction when the painful stimulus is applied during sleep, and the withdrawal of the limb.
- Group D2 includes those patients who have lost all facial and vocal reaction to pain, but who show a waking reaction when stimulated during sleep and can still withdraw a limb.
- In group D3 are included those whose only reaction to pain is withdrawal of a limb.
- D4 includes those patients who have lost all forms of motor reaction to pain.
Autonomic reaction (V)
These are assessed by watching the reactions of respiration, of the
ECG, and of the alteration in pupil size following painful stimulation.
In the majority of cases, a painful stimulus causes a period of apnoea
followed by a longer lasting tachypnoea. The heart rhythm may either ac
celerate or slow down. Quite often vasomotor changes are observed - rubor,
sweating. Mydriasis is also quite common.
This group is relatively simple, as autonomic response to pain is either
present or absent; only rarely have we noted dissociation of its components.
Group V1 includes those patients who react. Group V2 concerns those in
whom no autonomic reaction to pain can be demonstrated.
The end of the examination consists in testing the classical reflexes: the swallowing reflex, tendon reflexes, and cutaneous reflexes.
We would like to stress the importance of ob serving from day to day the changes in muscle tone. For this, it seemed to us that photographs or whole body diagrams could be of great use. We shall mention the three main groups most com monly encountered (Fig.7): the rigidity of de cortication with the upper limbs in triple flexion, the lower limbs in extension and a bilateral posi tive Babinski sign. This form of rigidity in man is sometimes accompanied by a positive Magnus De Klein reflex. In contradistinction to this, there is the very rare rigidity of decerebration (some times seen in children but hardly ever observed in adults for any length of time) with hypertonicity of the limbs and flexed wrists. And finally, there is the important group of those with no changes in muscle tone.
TABLE 2
Clinical form used for the assessment of nervous function in prolonged coma.
| Perceptivity (P) | Reading and execution of written orders | Orientation in time and space | Execution of spoken order | Blinking to threat |
| 1 | + | + | + | + |
| 2 | 0 | + | + | + |
| 3 | 0 | 0 | +/- | + |
| 4 | 0 | 0 | 0 | + |
| 5 | 0 | 0 | 0 | 0 |
| Aspecific reactivity (R) | Orientation reaction (awake) | Waking reaction (sleeping) |
| 1 | + | + |
| 2 | 0 | + |
| 3 | 0 | 0 |
| Motor reactivity to pain (D) | Facial mimic - Vocal reactivity | Waking reactivity | Limb withdrawal |
| 1 | + | + | + |
| 2 | 0 | + | + |
| 3 | 0 | 0 | + |
| 4 | 0 | 0 | 0 |
| Autonomic reactivity (V) | Respiratory variations | Vasomotor changes | Changes in cardiac rhythm | Changes in pupil size |
| 1 | + | + | + | + |
| 2 | 0 | 0 | 0 | 0 |
Tentative anatomoclinical classification
By correlating the preceding classification with anatomical observations on our own cases and others described in the literature, we have been able to distinguish four main anatomoclinical stages in prolonged coma (Fig.8).
The reactive apathic hypoperceptive syndrome
The word apathic is used here as the etymological opposite of hyperpathic. This syndrome corresponds to the one first described by Cairns et al. (1941) under the name of akinetic mutism. It concerns those subjects in whom perception is altered but not abolished (P3-P4). Reaction is normal (R l) and the orienting reaction is facilitated, hence the frequent ocular movements. Autonomic reac tions are normal but motor reactions to pain are partly lost. Even strong stimuli fail to elicit the characteristic facial mimic in response to pain. The lesions responsible for this syndrome spare the cortex but affect the thalamus (Cairns 1952; Jouvet and Dechaume 1961), the posterior part of the third ventricle; it can be a pineal tumour, a craniopharyngioma or an epidermoid cyst(Cairns 1952), occlusion of the vertebrobasilar system affecting the lower end of the brainstem but sparing most of the tegmentum (Kubik and Adams 1946; Cravioto et al. 1960; Girard et al. 1962). Finally, it can be a tumour of the cerebellum or of the posterior cranial fossa (Jefferson and Johnson 1950; Longman and Tenuto 1956; Daly and Love 1958). Of the cases reported of a kinetic mutism, only a few involve the cortex: cingular cortex (Nielsen and Jacobs 1951) and pallidum, by CO2 poisoning (Cravioto et al. 1960). It is also worth noting that in the majority of cases, the mesencephalic tegmentum and the reticular formation are virtually intact. It would appear therefore that the syndrome of akinetic mutism may result from a group of lesions primarily affecting the cingular cortex and the brain stem, damaging the extra pyramidal mechanisms concerned with the integration of motor responses to pain and to the spoken word.
The reactive hyperpathic-hypertonic aperceptivity syndrome
This is the equivalent of decortication in man. Perception is totally absent (P5) except if the occipItal. cortex is still intact, when the blink reaction may persist (P4). Reactivity is preserved (R1): Reactions to pain are normal, but, like the autonomic reactions, they may be exaggerated. There is always present a decortication rigidity with flexion of the upper limbs and frequently there is a positive Magnus-De Klein reflex. Finally, these subjects almost invariably keep their eyes open and appear awake all the time. Continuous recordings however show periodically a short lasting sleeping phase. The same signs were demonstrated in cases described by Rosenblath (1899) and by Kretschmer (1940) under the name "das appallische Syndrom", by Strich (1956,1957) under the name of "dementia from traumatic ence phalopathy", by Lundervold (1954) under the name of "loss of consciousness due to anoxia", by Zeman and Youngue (1957), Denst et al . (1958), Fischgold and Mathis (1959), Nystrom (1960), and by ourselves (Jouvet and Dechaume 1960; Trillet 1961).
In all the cases, the lesion is similar. There is either massive cortical involvement (Denst et al. 1958) or diffuse degenerative changes in the white matter of the cerebral hemispheres. The brainstem is intact in the majority of cases (with the exception of the pyramidal fibres which frequently show signs of degeneration).
The areactive apathic normotonic aperceptivity syndrome
This group concerns cases of very deep coma in which survival is usually limited to a few weeks. Perceptivity is lost (P5). Nonspecific reactions are altered (R2-R3) as well as reaction to pain (D2-D3), but autonomic reactions are normal. In most cases, there is no definite hypertonicity. Patients described by Jefferson (1952) under the name of parasomnia, by French (1952) under the name of prolonged uncounsciousness, and by Cravioto et al. (1958), and Trillet (1961), present the same symptomatology. The lesion common to all these patients is one affecting the upper part of the brainstem (mesencephalic reticular formation), but in most cases, there were also associated lesions of the cortex or white matter. It appears that total or subtotal destruction of the reticular system is responsible for the more severe losses of reactivity.
The aperceptivity areactive apathic and atonic syndrome
This syndrome, described only recently, is a result of modern resuscitation methods. In spite of diffuse damage to the encephalon (most often from anoxia) and of the development of massive aseptic necrosis, life can be maintained by arti ficial respiration and by continuous perfusion of hypertensive agents. This group consists of the cases of brain death (Wertheimer et al. 1959), sometimes called "coma dépassé" (Mollaret et al. 1959) or artificial survival (Bertrand et al. 1959). Such a clinical picture poses the problem of legal death (which is discussed elsewhere). We shall only mention, as a reminder, the various tests used to diagnose death ofthe nervous system and on the results of which one is entitled to stop artificial respiration (Fig.9):
- no spontaneous resumption of respiration when the respirator is switched off
- absence of recordable cortical and subcortical electrical activity from a deep electrode
- poikilothermia
- polyuria
- failure to fill the cerebral vascular tree by arteriography.
Figure 1 : Diagram of sagittal section through the brain of a cat
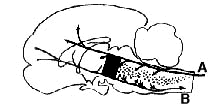
In black: mesencephalic activating reticular system responsible
for waking.
Dotted area: Raphé system responsible for sleep.
(A) specific afferent pathways;
(B) motor efferent pathways.
Figure 2 : Disappearance of decortication rigidity during sleep
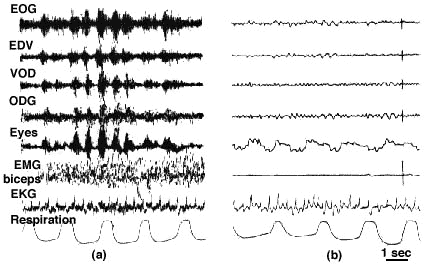
(A) Waking state (eyes open) in a patient pre senting a chronic syndrome of decortication for several months. Note the numerous muscular artefacts on the EEG leads, as well as the high muscle activity in the biceps.
(B) Disappearance of muscle activity during a period of paradoxical sleep characterized by the rapid eye movements (4th line).
Calibration: 1 sec 50 microV
Figure 3 : Integrity of the lower brainstem is not essential for consciousness
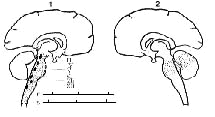
(1) Dotted area: extent of cranial nerves paralyzed after intravertebral
injection of 50 mg of AmobarbItal. Despite extensive involvement of the
brainstem the subject is still able to respond (R) to a sensory stimulus.
See details in text.
After Rossi (1965).
(2) Diagrammatic representation of a pontine lesion (by softening)
resulting in decerebration rigidity with preservation of normal perceptivity
(still able to open and shut eyes wken ordered).
(Halsey andDownie 1966.)
Figure 4 : Traumatic encephalopathy
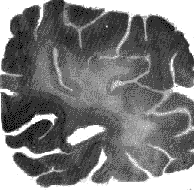
Extensive demyelination of white matter in the occipItal. lobe (Woelcke's
stain).
(Trillet 1961.)
Figure 5 : Massive aseptic necrosis in brain death
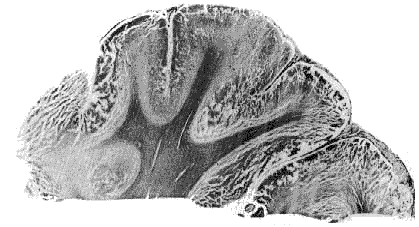
Frontal cortex : ex ensive necrosis with fissuring of cortex.
White matter is fairly well preserved.
Hemalum, phloxine, saffron. (Trillet 1966.)
Figure 6 : Hyperaesthesia in a decortication syndrome
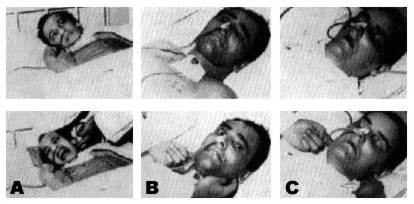
(a) Hyperaesthesia in a decortication syndrome.
(b) Absence of facial mimic, but norn-al arousal reaction (type
D2).
(c) Absence of mimic; loss of waking reaction. See text for details.
Figure 7 : Decortication rigidity
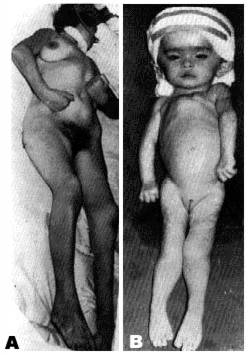
(a) Decortication rigidity.
(b) Decerebration rigidity.
Figure 8 : Diagrammatic anatomoclinical correlation of main forms of coma
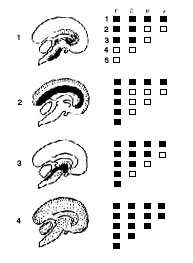
On the left: the most common lesions are shown in black on a
sagittal section of the encephalon; less frequent lesions shown as dotted
areas.
On the right: results of the clinical examination. Black squares
indicate a negative response to each test.
- (1) Akinetic mutism: P3-D3-R1-VI.
- (2) Decortication syndrome: P5-D1-RI-VI.
- (3) Upper brainstem lesion: P5-D3-R2 Vl.
- (4) Brain death: P5-D4-R3-V2. See details in text.
Figure 9 : Failure to fill the cerebral vascular arteries by arteriography in brain death
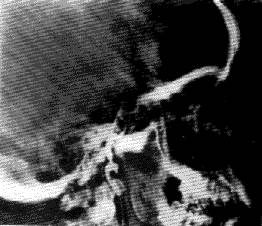
The contrast medium enters the internal carotid, but does not reach the distal part of the carotid artery system.